
Published: August 18th, 2017 Last Modified: February 16th, 2019
Depending on the type of work you do, there may be enzymes that you go through like grad students with free cookies at seminars. For us, Q5 DNA polymerase and Superscript reverse transcriptase are two that are indispensable. Thing is, I actually don’t think these enzymes are horribly overpriced for what they are. Both enzymes are the results of years of mutagenesis and testing and are at the bleeding edge of polymerase technology. New England Biolabs, especially, charges a reasonable amount for their enzymes, Q5 included. On top of that, you can easily dilute Q5 by 1:2 to 1:4 and achieve reliable results while Superscript can be diluted to 1:8 (25U/rxn!).
Despite the reasonable prices, there are times when I think using high end, store bought enzymes are an absolute waste. You are not only paying for a fellow pipette jockey to purify the enzyme to homogeneity, but also for extensive QC and packaging. So when you take such a beauty of an enzyme and dump it into your colony PCR, well, I shed a tear no matter where you are in the world, I can sense it. I would like to see manufacturers sell two “grades” of enzymes, one that is ultra-pure for industrial/commercial users who need absolute traceability, and a lower grade for basic research use. I’m not holding my breath though, which is why we’ve spent time purifying the best equivalents to Q5 polymerase and reverse transcriptase (RT) I could get my grubby little hands on. Combined, we’ve amassed enough enzyme to last decades with a Molecular Biology Black Market (TM) value over $150,000!!! In this first post I will cover DIY pfu-sso7d and DIY reverse transcriptase will be next.
Unmodified, stock Taq DNA polymerase has been the standby choice of PCR for decades now, however as the demands for PCR have increased it has been shown to be lacking in both processivity (nucleotides/second), fidelity (errors in sequence) and resistance to various inhibitors.
Superior thermostable polymerases from other organisms have been isolated (Pwo, KOD), however Pfu from Pyrococcus furiosus came out on top as the popular upgrade to Taq, with a 10-20 times lower error rate vs Taq, depending on who you ask. Targeted mutagenesis as well as blending complementary enzymes have further enhanced earlier commercial offerings. The most recent development of the art have involved creating domain chimeras of polymerases (KOD + Pfu = Kofu, Pfu + KOD = POD, cute eh?) as well as fusing DNA binding domains (sso7d) to these enzymes. For more detail as to these developments have a read through this nice review.
It’s worth noting that plasmids for Taq (pAkTaq) and Pfu (pet16b-pfu) are out there, however when a superior enzyme with an identical purification protocol is out there, it really isn’t worth going for anything less.
These days, the all-round best PCR enzymes you can get commercially are Pfu-sso7d variants sold by the usual suspects, with 30-50x the fidelity of Taq. NEB’s Q5 polymerase represents the bleeding edge and boasts 100-200X fidelity. Ideally, we would want to purify our own Q5 enzyme, however NEB is understandably tight lipped as to what Q5 actually is. NEB’s latest patents don’t give too much away, however we can atleast glean that Q5 has exonuclease activity and is fused to a sso7D domain. At this point I’m not sure if it’s a super mutagenized pfu-sso7D variant or perhaps a Kofu-sso7d. I will update this post if I figure it out, but basically if you could find the right patent and determine the nucleotide sequence, then a clone of Q5 would cost you less than 1000$ via gene synthesis.
Can we get the second best enzyme, pfu-sso7d, sold commercially as Phusion? YASSSSS!!! The Barrick lab, at UT Austin, has the plasmid, sequence, and isolation protocol available to academic users.
*****Update: 16/09/18*****
One note, this is just wild type pfu fused to wild type sso7d. Most commercial variants have mutations which increase various properties of the enzyme, like fidelity and processivity. Better variants of sso7d exist as well. However, stock pfu-sso7d is a respectable enzyme by itself, and would serve as a good base to build on if you were into mutagenesis.
Thank you to the Magos who gave me insight about pfu-sso7d! They also gave us the recipe of their version of 5X pfu-sso7d buffer! (See below)
************************
After obtaining the plasmid the isolation protocol was your usual fare of induction, lysis with a sonicator, heating to denature E. coli proteins and passing through a nickle column. For those of you unfamiliar with protein purification, don’t let the nickle column throw you off, it’s easier than you think.
Instead of a dialysis cartridge we used a concentration column with a 30 kDa cutoff. For quality control, we compared the activity of the DIY-Phusion vs commercial enzyme from NEB to determine the units/uL. As well, we performed assays to determine any residual nuclease activity as per the NEB datasheets for phusion.
Basic QC Tests:
- Do a PCR for a few reasonably sized products with a gradient of purified phusion vs NEB’s phusion. Run on agarose and compare intensities, nothing fancy. We chose to purchase a vial of Phusion for 120$ or so. If you didn’t want to buy a tube, you could always just add arbitrary amounts to set volumes, like 0.5 uL of enzyme/50 uL rxn volume.
- Incubate some good quality plasmid DNA/total RNA with differing amounts of your enzyme, run on a gel and compare against fresh plasmid/RNA. Degradation indicates some level of nuclease activity. For NEB, <10% degradation after 4 hours of 37C or 72C is good enough.
You can either buy NEB’s 5x Phusion buffer pack or make your own buffer, see recipe below. The homebrew buffer needs full validation, so if you make it please let me know of how it went, what template/product you used etc. The NEB buffer pack (6×1.5mL?) is 25 bucks or so, honestly it’s not a bad deal, however from my testing the homemade buffer seems to perform better in my hands (Proof below).
From a single purification we managed to obtain ~100,000$ worth of Phusion polymerase, which will last us for some time. While DIY-Phusion won’t replace commercial Q5 in our lab, it allows us to save it for when such a high fidelity enzyme is called for. For routine PCR under 7 kb, phusion is an absolute workhorse.
Sounds great but what about costs? Depending what you have in your fridge, you need 50-200$ worth of nickle beads, 20-100$ for dialysis and concentration columns and some method of lysing your cells. Don’t forget to factor in your time as well (How much is an hour of your time worth? Hmm…). Budget 2 days for purification and maybe 2-3 for validation and quality control.
Hope that inspires some of you to purify your own enzymes and lessen your dependence on commercial suppliers.
Here are 5X pfu-sso7d buffers I’m testing to provide an alternative to NEBs buffer pack. Will post proof/gel pictures as I get them.
5X HF Phusion Buffer Recipe 1 (Source)
150 mM Tris-HCl pH 10
50 mM KCl
50 mM NH4OAc
10 mM MgSO4
0.5% Triton X-100
0.5 mg/mL BSA
Proof (800bp): Tried homemade buffer #1 vs storebought NEB 5X HF buffer with homemade phusion, PCR for mTAG-BFP2 from ~25 ng plasmid. Overall it worked, if I’m being picky there seems to be a bit more smearing in the homemade buffer, however the amount of desired product is the same between the two buffers. Is it a difference in buffer quality or is it my handling? Will try a larger, more challenging template next.
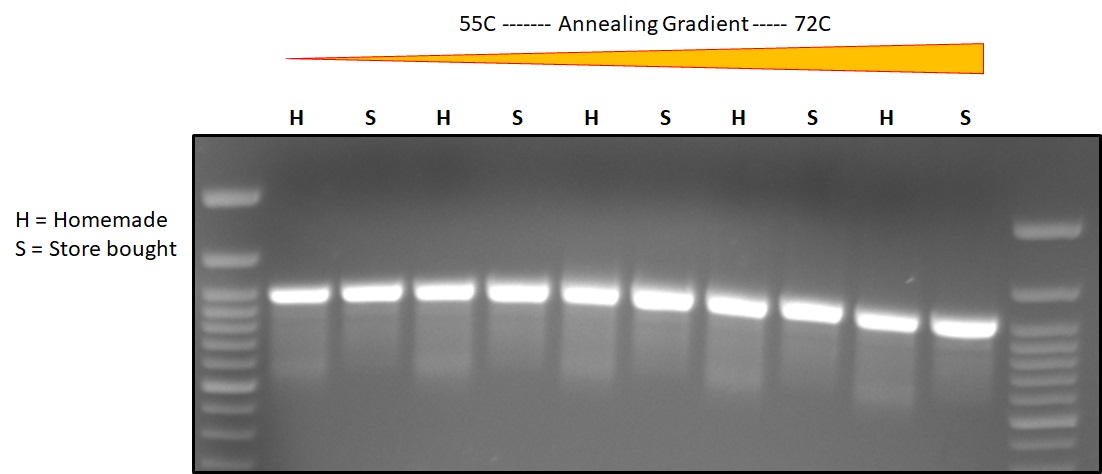{kind=link}
Proof (5.5 kb): Playing around with GC buffers to amplify difficult samples. Not much luck in that department, but as you can see from the gels that our homemade buffer actually outperforms NEBs HF and GC buffer. Template was a 5.5 kb vector to be used for Gibson assembly of Mashup-RT 😉
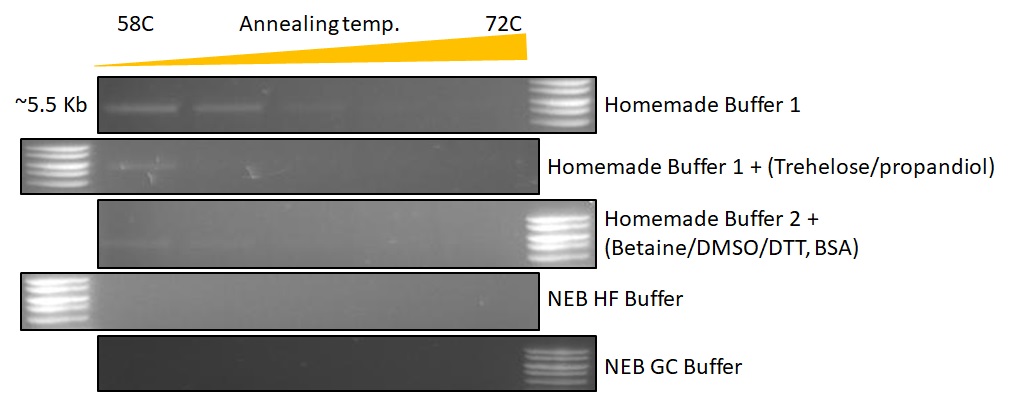{kind=link}
Proof (9.5 kb): Looks like our humble phusion can do 9.5 kb of a viral genome without any GC enhancer (albiet not very much product). Still working on homemade GC enhancer buffer. Q5 without GC buffer is better, but dependent on primer choice. Q5 + GC enhancer absolutely dominates, couldn’t work with my potyviral clones without it.
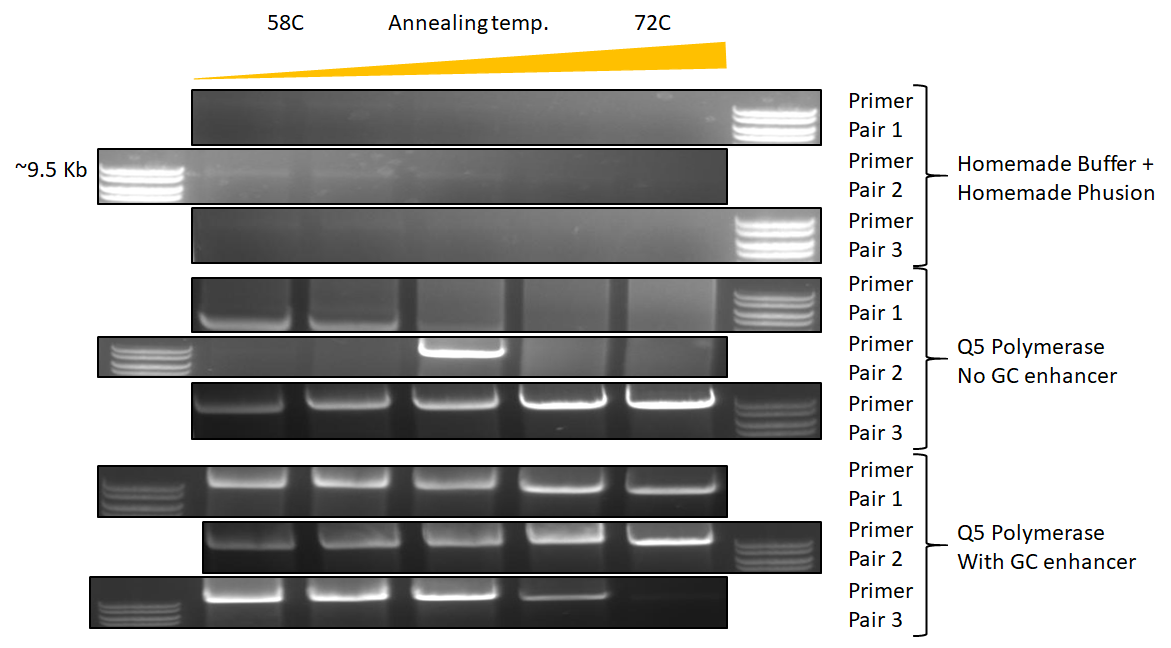{kind=link}
5X HF Phusion Buffer Recipe 2 (Source)
150 mM Tris-HCl pH 10
200 mM K2SO4
5 mM (NH4)2SO4
10 mM MgSO4
0.5% Triton X-100
Proof: Had to order potassium sulfate, didn’t have it on the shelf. Made up Buffer 2 with and without the amount of BSA present in Buffer 1. Tested against NEBs HF buffer and the successful Buffer 1 from last time. Amplified eGFP from a 16 kb viral plasmid, (-) indicates viral plasmid without eGFP and (+) indicates with eGFP, used ~25 ng plasmid. Interestingly only the homemade Buffer 1 actually managed to get the desired product. It’s possible that the source of plasmid (Agrobacterium silica midi-prep) is a difficult template, the quality of plasmid you get from agro isn’t as good as E.coli (but still better than yeast plasmid preps). Potentially I forgot to add an important PCR component, but It’s unlikely I did it for 3/4 mastermixes. Not ruling out Buffer 2 out of the race just yet, but looks like Buffer 1 is the current winner. Will retry with an E. coli plasmid as template and will show amplification of 3 and 9 kb fragments.
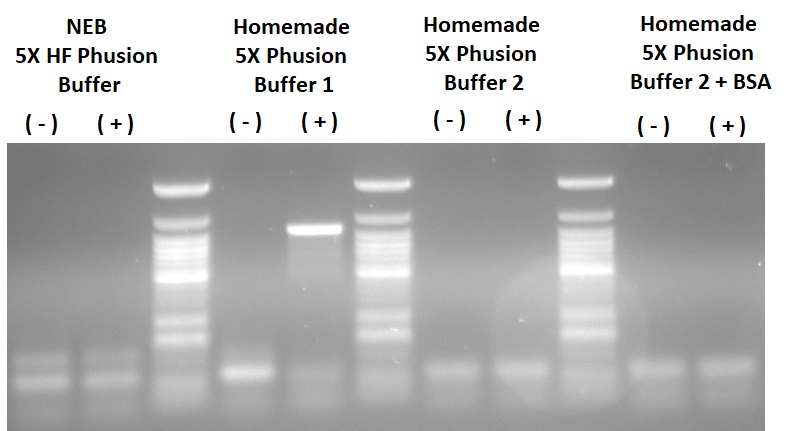{kind=link}
5X HF Phusion Buffer Recipe 3 (Full credit to a fellow Magos Biologis, Source1, Source2)
1000mM Trehalose
500mM Sarcosine
400mM KCl
80mM (NH4)2SO4
50mM Arginine
50mM POPSO
7.5mM MgCl2
0.5% Tween-20
5(-25%) Glycerol (depending on GC)
5(-15%) DMSO (depending on GC)
pH = 8.8
Additional components for Green Buffer (for direct Gel loading);
<12.5% (w/v) Ficoll 400 (as low as possible whilst still allowing for crisp bands)
0.03% (w/v) Tartrazine
0.003% (w/v) Xylene Cyanol
Thank you for sending this one in, let me know if you want a link to your stuff. Will try with stock pfu-sso7d and some decent sized templates.
*********************************
Regarding homebrew reverse transcriptase, check out MashUp-RT!
If you are an academic user and would like to try the plasmid, contact me. If you would like to add to the conversation so that everyone can see and benefit, leave a comment/feedback in the comments at the very bottom of the page.
Hi Pipette Jockey,
This is a very nice post !!!! Any Chance you have figure out Q5 yet? I am tempted to synthesized
Kofu-sso7d, but i’m not sure what kind of buffer it will work in… Also It will be nice if you can compare notes with me regarding the sequence of Kofu. let me know.
The identity of Q5 seems pretty elusive, I’m not sure if we will ever know. The thing about patents is that while they do give you a monopoly to produce something for a given length of time, it also reveals to the world exactly what your secret sauce is, which is why it’s understandable there’s very little known about it other than its a “novel” polymerase with sso7d.
As for Kofu-sso7d, I know the temptation, It seem pretty promising. However unfortunately the patent does not give much actual data about the error rates of these chimeras, so it’s impossible to say that Kofu-sso7d would be superior to pfu-sso7d. Moreover, in the same patent they produced Kofu-II and Kofu-III, which one to chose?
So, with that many unknowns it’s hard for me to recommend anyone to go and get Kofu-sso7d synthesized because it’s real money we are talking about, probably about 1000$ or so per clone, on top of reagents and your time to purify it and optimize the reaction conditions (weeks?). On top of that, any work you do won’t be publishable or marketable. And that’s time and money that can go into your own publications, you know? That’s why we ended up going with homebrew phusion, it has very good characteristics with a minimum of time invested.
Now, if you have money and time a-plenty, the patent states that for kofu they used Novagen’s KOD buffer 1, which could be derived from this paper:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC168769/
“The buffer components were 120 mM Tris-HCl (pH 8.0), 1.2 mM MgCl2, 6 mM (NH4)2SO4, 10 mM KCl, 0.1% Triton X-100, and 0.01% BSA for KOD DNA polymerase”
You are correct, from the patent, Kofu have the same fidelity as wt-Pfu but hive higher processivity. However comparison to Pfu-sso7d is not available. Can you kindly shoot me a personal email. I wanted to ask you some tips of isolating phusion. 🙂
Kofu-sso7d might be something akin to Thermo’s Phusion-Flash enzyme, which has incredible processivity (a la KOD) and Pfu-like error rates. This is a very useful enzyme (PCR reactions <1 HR)
With the cost of synthesis these days it’s tempting to give it a try. You’d have to keep it to yourself but there’s nothing stopping most researchers dropping about 500 bucks for synthesis to get something like kofu-sso7. What I think we will see is fewer companies actually patenting their stuff for this reason.
I will revisit pfu-sso7d at some point, either with mutagenesis or going for a different chassis altogether.
I have good experience with PfuX7 used for USER cloning. It is a Sso7d fusion polymerases with a specific mutation so it does not stall when in encounters a uracil. In two days, I produced enough enzyme to last the whole of my PhD!
This article describes the cloning of PfuX7 and comes with a complete protocol for purification, PCR running conditions and buffer composition.
https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-10-21
That’s a really cool paper, thank you! Jeese, I thought I’ve tried every cloning method under the sun , I’ll have to ask for the plasmid and give it a go!
I’d love to do some random mutagenesis on the Pfu-sso7d plasmid, to get it up to snuff of NEB’s Phusion, that would be fun 🙂
I’ve been working on purifying pfu-sso7d (which my lab obtained from the Barrick Lab, as you mentioned), but I’ve run into a problem with the Phusion losing its activity very quickly. Following purification, I used dialysis to put the Phusion enzyme in the buffer recommended by the Barrick Lab (http://barricklab.org/twiki/bin/view/Lab/ProtocolsReagentsPfuSso7d). However, within a month, Phusion had lost all of its activity. I tested it for aggregation and degradation, but the results didn’t show much. Did you run into similar issues?
Hmm, so far I haven’t noticed any significant loss of activity. How did you test for aggregation and degradation, running it on a SDS-PAGE gel? What did it look like? Let me know, could help with troubleshooting. We used a 30 kDa column to get rid of any imidizole in the elution buffer then put through the storage buffer several times. The storage buffer was made with fresh DTT. After that, I still noticed that the protein was freezing in the -20C, so I added a touch of glycerol until it stayed liquid. I also aliqouted the phusion into 100 uL portions to limit how much time they spent out of the freezer.
One thing I’ve noticed is that you need to tune in the units/uL of phusion you use reasonably well, I was finding that too much enzyme inhibited my PCRs.
If you take a look at NEB’s storage buffer, it’s got some BSA in it, presumably to stop the phusion from sticking to the tube in storage, wouldn’t hurt to add some for flavor. Also, there’s “1X stablizers”, not sure what that entails but I’ll try to dig through the patents to find out.
Losing of activity is due to NP40. If you don’t use a appropriate grade NP40, polymerase will lose its activity within a month or so. I have purchased NP40 from milipore and that worked quiet fine. We are still using 5 years old enzyme now. Hope that helps.
Thank you for the advice! Our last batch had a shelf life of about two years before the activity took a nose dive. The freezer dying during the great freezer debacle of 2019 could have been a contributor. At any rate, we use Sigma branded stuff, I believe detergents are one of their specialties.
I am facing the same issue with the commercial phusion polymerase. Can you help me out in this regard. Did you find any solution to reactivate/stabilize the enzyme. Thank you.
I have recently procured a Pfu-sso7d plasmid from someone (whom I will not name publicly) recently and I am planning on expressing the enzyme. They use a different expression protocol…
1) Induce 6L of Phusion for 5 hr at 37°C @ OD600=0.6. Clone is Kan resistant in pET28. Express in BL21[DE3]pLysS
2) Harvest and wash once in PBS
3) Resuspend pellet in 100 ml
50mM Tris pH 7.9
50 mM Dextrose
1 mM EDTA
1 mM PMSF
5 mM B-me
4) Freeze in liquid nitrogen
5) Thaw cells and add 100ml of same buffer plus lysozyme to a final concentration of 1mg/ml, stir until well resuspended and lysing. Sonicate to help things along.
6) Add an equal volume of lysis buffer (200ml) and mix well
10mM Tris pH 7.9
50 mM KCl
1 mM EDTA
0.5 mM PMSF
0.5% Tween-20
0.5% Igepal CA630
5 mM B-me
7) Heat at 75°C for 60 minutes then cool on ice.
8) Spin at 10,000 RPM for 20 ‘ in a GSA rotor
9) Measure supernatant volume and add 3M KCl to a final concentration of 100 mM
10) Filter through a 0.45 µm filter.
11) Load onto a preequilibrated 50-75ml Bio-Rex 70 column
12) Wash with 2 CV of 100 mM KCl in lysis buffer then 2 CV of 200 mM KCl in lysis buffer.
13) Elute with a 200-500 mM KCl gradient. Pool and dialyze into storage buffer with 50% glycerol to store at -20°C.
And when I look at commercial storage buffers, I see something called “stabilizers”. Like this…
Supplied in: 20 mM Tris-HCl (pH 7.4 @ 25°C), 0.1 mM EDTA, 1 mM DTT, 100 mM KCl, stabilizers, 200 µg/ml BSA and 50% glycerol
I have seen this webpage “https://www.quora.com/Though-taq-polymerase-is-thermostable-it-is-advised-that-it-must-be-stored-at-0-to-20-degree-Celsius-Why-is-it-so” that points out a patent which use Arginine as a stabilizer. Ever tried that?
Hey buddy,
I’ve thinking about what “stabilizers” are in some of NEBs buffers too since someone mentioned they were having stability problems with their phusion. I haven’t tried playing with different preservatives because atleast for now, the batch of enzyme I made appears to be working a year or more later stored at -20C. Keep in mind I used a concentration column to clean/concentrate my protein instead of dialysis, so I had to add some extra glycerol to the final protein to keep it from freezing in the -20C. The Barrick protocol uses NP-40 and Tween-20 in their storage buffer, which is similar to the detergents used here (https://patents.google.com/patent/US7972828B2/en), and according to their testing these detergents preserve polymerases for quite some time. If you take a look at the buffers for NEBs older polymerases, they have detergents, but not “stabilizers”.
I’m thinking when they started to develop detergent free buffers (for whatever reason, can’t quite remember why you don’t want detergents for some PCRs…), they started playing with other additives. It would be interesting to try the arginine you mentioned, as well as some sort of phospholipids (https://patents.google.com/patent/US20120282669), but yeah, for relatively routine PCRs the Tween20/Igepal630 storage buffer seems stable and works.
Thanks for the nice bit of information 🙂
Looking through several purification schemes I’d like to bounce a couple of ideas upon the Barrick protocol…
1- High temperature incubation – I will definitely add this step for extra clarification.
2- PEI precipitation of DNA – One thing I don’t know is whether the protein crashes at high salt… My experience from helicases during PhD was that some DNA binding proteins would be happy even in very high salt concentrations (I went up to 2M) during purification to get rid of waste DNA.
3- No reducing agent in buffers? – Most of the procedures concerning Pfu polymerase include a heavy amount of B-me (or DTT) in the purification buffers. I’d probably include that in the buffer mixes.
Looking at what is there in my lab (currently, as a PI, I am not doing protein science) the only thing I have is Ni-NTA so that’s why I cannot use ion-exchange or size-exclusion, so Barrick protocol makes the most sense. Do you have any off-the-record modifications you did to the Barrick protocol? I’d really like to hear those.
Take care, MadLab
p.s. Btw really digging the gel electrophoresis apparatus 🙂
Sorry to get back to you so late, not sure why I didn’t get a notification. The Barrick protocol has a 70C incubation for clarification, but I think you could crank that a bit higher, albeit for less time (I think some Taq protocols do this). As for off the record stuff, hmm, like I mentioned, we used a 30 kDa for purification/concentration instead of dialysis, and we added just enough glycerol after purification to stop the enzyme freezing at -20C. The cells we used for expression were Rosetta 2 (DE3) pLysS. Apart from that, I don’t recall anything unusual…we also did Ni-NTA resin, was reasonably fresh stuff. Yeah, I don’t like keeping secrets from you guys, I’ll tell you everything! Check out the two new putative buffers I posted, looks like I won’t have to buy any more NEB buffer packs!
I’m glad you like the gel box/combs 🙂 It’s got that old school cool, which I appreciate. PEI combs are the way to go for regular gels, and PTFE is nice and slippery for low-melt agarose where the wells tend to rip.
1) Essential purification step for thermostable polymerases. This alone yields pure enough enzyme for use out of the gate. However, residual plasmid DNA has the potential to mess up your reactions.
2) I do not recommend this. PEI will do a terrific job of precipitating NA but residual PEI is quite difficult to get rid of. You’ll want to perform it in high salt (~1M) to ensure your polymerase doesn’t precipitate with the salt. Typically labs follow this with an ammonium sulfate precipitation of the protein of interest, but this comes with its own problems.
3) You can wash your Ni-NTA bound protein with high salt (1M) and this will get rid of a lot of contaminating nucleic acid, but not all of it. I recommend this if you really have no access to ion exchange resin.
You should really poke around for ion exchange resin. Many old (>20 years) biochemistry labs will have dry DEAE or CM resin lying around. The stuff was relatively cheap in the old days. Either will substantially increase the purity of your Pfu-Sso7d albeit in different ways.
Great Post and Blog!
Invaluable DIY resource for scientist in non-central countries.
Could it be possible to get help on getting the plasmid?
Looking forward for the superscript chapter….any teaser available?
Keep posting!
Thanks! I’m glad you find the blog useful 🙂
I’ll email you about sending the phusion plasmid, the creator was very generous with his redistribution clause.
The superscript is a bit more complicated…we actually have a clone of reverse transcriptase that’s the equivalent of Superscript 3, however we do not have permission to redistribute it and out of respect I’d rather not mention them directly on the blog. With that said, if you read the latest literature on reverse transcriptases and ask nicely, you can get your hands on some awesome enzymes.
Thanks for the answer!
Regarding the reverse transcriptase, seems to me I should run a Marathon then…..let me know if I’m wrong…
TNXS again!
Thank you very much!!
Thank you very much for your posts. These are very helpful for scientific community from developing countries where funding for research is scarce.
In our cloning projects, Q5 has consistently outperformed Phusion. To decipher the mystery of Q5, one might use nested PCR to amplify trace amount of DNA plasmid present in this enzyme. To get enough templates for PCR, DNA could be concentrated from Q5 (100-500 µl) by ultrafiltration. If you have sequences of different versions of Kofu, we can give it a try.
In the meantime, please let me know how to proceed to get the Phusion plasmid.
Hmm, maybe mass spec for Q5 and KoFu? 🙂
If you can recommend a few places that can sequence whole proteins I would love that. I looked into protein sequencing a while back and it seems most people sequence the ends, whereas something over a few dozen amino acids becomes an interesting challenge.
Do you know sequences of random hexamers for C-DNA synthesis
Well, in theory they should be random sequences, however it’s possible that during their synthesis there ends up being a bias towards certain sequences. Not sure if anyone has done a study on the actual sequences of random hexamers, but there is plenty on what biases can arise from using them for cDNA synthesis.
Thank you for your information
Anyone knows RNA lysis buffer composition ( RLT) for spin coloum isolaton
I don’t personally use the RNeasy kits, but I seem to have found something that could be useful for you, take a look at this patent:
https://patents.google.com/patent/US20140356860
Just taking a look at the RNeasy protocol, it seems you have to add an equal volume of 70% ethanol, which brings it in the 35% range, which is close to what they talk about ” The resulting binding mixture comprised the chaotropic salts GTC and GuHCL in an overall concentration of 2M, 33% ethanol and Triton X-100.”
This is a bit of a guess, but here’s what I would try first:
Putative DIY Buffer RLT:
4M Guanidine Thiocyante
10% Triton X-100
can we use Phusion emzyme for QPCR like using Evagreen or SYBR dyes
Assuming you could buy the sybr dye separately and went through the effort of making your own mastermix, absolutely.
You can buy SYBR in bulk from China (but ask for samples first, also be on the lookout for batch-to-batch variations). It comes 1/10 price of regular SYBR. I would advice to use Taq while doing the optimization instead of Phusion because Taq seems more robust and you don’t need accuracy in qPCR, only comparability and robustness. But check against a commercial kit first.
Also optimization is a bitch…
Thanks for all your insight as usual, I appreciate it. I sent you an email to keep in touch.
I have a LOT of qPCRs coming up, and honestly it could be a good idea to make my own blend. But yeah, optimization IS a bitch. With all these cost saving measures you always have to factor in the worth of an hour of your time, how much labor you have VS just buying a tube of pre-made stuff.
Dear Pipette Jockey,
Thank you so much for your post and efforts in helping people around.
I wonder if you ever tried to establish the vector and purify reverse transcriptase? For people who need to do a lot of one-by-one cDNA synthesis that would be a really helpful tool.
Hello my slavic brethren 🙂
I’m actively developing a RT plasmid I can freely distribute to labs, I have all the juiciest mutations figured out. The only thing holding me back from getting the plasmid synthesized immediately is concerns about patents. Most/all of the good mutations in MMLV-RT have been patented, so I’m in talks with lawyers to see what is possible without getting sent straight to the gulags.
But yeah, a RT is at the top of my list, I’m pretty obsessed with the project. I’ll make a post as soon as it’s available.
Dear Pipette Jockey,
This is quite invaluable. What sort of yields are you getting for the Phusion polymerase per liter of cell culture grown?
Tim
Hey,
I’ll get more detailed numbers for you, but from 400 mL of induced culture we obtained about 100,000 units worth of the enzyme. You shouldn’t have to do the purification too often 🙂
how your calculating unit activity , any simple assay?
Basically I am calculating activity relative to a gradient of store bought phusion polymerase. So, do a PCR for your favorite template with a gradient of store bought pfu-sso7d and another with a gradient of your purified enzyme, run them both on a gel and compare them. That’s the simple way, if you wanted to do it by the books you would follow NEBs protocol, I think they measure phosphate release or something like that.
The mystery of Q5 is ever elusive… Isn’t there someone reading here who has access to a protein sequencing pipeline (Mass-spec) and could just fire an aliquot of Q5 through? ^^
Best I have for you guys so far is that kofu-sso7d works in Q5 buffer and not in phusion buffer, take that as you will.
Yeah protein sequencing would be cool, but it’s a non-trivial experiment, probably 500-1500$ last time I took a peek at prices.
Then you gotta optimize the buffer, they’re not forthcoming about the recipe.
what is gc buffer composition is it betaine or DMSO?
Good question, it’s something I’m actively looking into. Unfortunately it’s not a patented mix, so it may come down to just optimizing your own homebrew buffer, start with the regular buffer and add DMSO, betaine etc.
Check out this paper, they made a 5X GC enhancer with Betaine, DTT, DMSO and BSA http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.553.4058&rep=rep1&type=pdf
betaine is very hard to dissolve in water ….. In this paper they made 5x Enhancer buffer which is impossible to make at room temperature.
Hi Alex,
Quick question – For the Buffer 1 recipe, should the final pH be 10 or just the pH of the TRIS? If that is the case I will also pH the NH4COOH.
Take care,
Hello! Usually I just use a stock of Tris pH 10. There is probably plenty of room for improvement with the buffer though, I still haven’t tried recipe #3 with the trehelose.
In case you don’t want to sell a kidney to be able to buy trehalose from Sigma then you can just buy it from any health and supplement store. The trehalose they have works equally well and is about 1000x cheaper! The brand I use is Swanson Ultra.
Hahaha, its too late for me, my first order was from Sigma at 500mg for 80$…These days we get it from Bio-Shop, and they sell it nice and cheap.
Thanks for the tip, I did wonder whether food grade trehalose would do the business, good to know! I’m not sure if they still do this, but there were spice suppliers that would sell 5 pound bags of silicon dioxide…who knew we all eat ground glass on a daily basis!
First Trial…
I have tried Buffer 1 (after looking at your faint bands, a faint band is all I need!) with my pain-in-the-neck PCR problem -> No product what so ever (usually what I see with commercial Phusion buffer is at least some kind of primer shenanegans – even haven’t seen that…)
Will give a second round…
Take care,
ML
Good post, thank you. I waste a lot of time to search for the storage buffer composition of Q5 enzyme. And finally I could not find it, but I find your blog, so it worth the time. 🙂 You mentioned at the beginning of the post, that you dilute Q5. Did you use it immediatelly or is it able to store after dilution? What kind of buffer did you use for dilution?
Thanks,
psari
Hey! So I dilute it by factoring in a smaller number per reaction in my mastermix, so instead of 0.5uL/50uL rxn I use 0.25 uL and make up the difference with water in the mastermix. If you wanted to dilute it separately I’d use water and then add it to your reactions immediately, I doubt it would store well diluted long term.
Great post, your blog is awesome!
Could it be possible to send the plasmid to a eastern european academic institution?
Thank you! I sent you an email before, maybe you didn’t get it? I’ll send you another.
Hey MADLAB,
Are you still there?
I would like to know if your BL21 (DE3) Plyss was codon optimized? Great name by the way.
Hey Pipettejockey,
Thanks for the effort you put in this website, it’s great to know that there’s a community out there. I would like to know if the expression of pfusso7D requires codon optimized strain. Same question.
I read MadLAb asking the question about NaCl concentration to get rid of DNA. In my purification I used 0.5M Nacl in my lysis buffer and 1M NaCl in my washing buffer. I would like to know your opinion on whether this concentration would precipitate the protein too.
Another question is: NiNTA is compatible with up to 100mM tris, my buffers contain 25mM tris pH8; I saw MadLab using 50mM tris buffer so I assume it should be fine.
Also thumbs up for a future SDS-page picture.
That’s it, glad for the community.
Hey, glad you enjoy some of the stuff 🙂
I blasted the pfu-sso7d from Barrick’s sequence on NCBI, it’s a 99.9% match with the wild type, so I don’t think it has been modified in any way. There’s plenty of performance to be squeezed out of the plasmid, for sure.
We used the Rosetta 2 DE3s that he mentioned in the purification, we happened to have it in the fridge too, so maybe that’s what it’s a case of.
Sorry, I personally haven’t played with the purification too much, we isolated a huge batch maybe two years ago and we haven’t gone through it again since. We followed the Barrick protocol except that we used 30 kDa Millipore columns to concentrate/buffer exchange the enzyme. Barrick uses 50 mM phosphate buffer pH 8.0, so 25-50mM Tris sounds alright.
Getting some help with the protein purification around here, and we’ll be making a fresh batch. I’ll take some pictures of the purification.
I don’t think my construct is also codon optimized but I get a shitload of protein for each run with standard BL21(DE3) – enough for +5 years for a lab which is doing PCR regularly.
Btw, I use PEI precipitation under high salt to get rid of excess DNA, or sometime do a DEAE-column after heat treatment – DNA gets stuck to the DEAE-column. Either way you get a low amount of DNA contamination.
I am still getting the DNA contamination after the DEAE column and PEI precipitation. Can you please tell me the the PEI concentration.
Greeetings,
I haven’t checked if the construct was codon optimized or not, however I teach Biochem and in the Biochem lab, I have made undergrad students produce Pfu-sso7d using regular BL21(DE3) and at the end of the lab session I had so much (and active) enzyme that I still don’t know what to do with it 😀
Hope this helps,
MadLab
Regarding Kofu, I am not sure I understand its benefit.
It has the speed, inhibitor resistance and processivity of KOD, but fidelity of Pfu. Pfu is less fidelous than KOD, so Pfu is generally inferior to KOD in pretty much every way.
Unless I am mislead…
In any case, this would be of more interest imho, http://ncbi.nlm.nih.gov/pmc/articles/PMC4489709/#pone.0131757.s001. they make KOD-Sso7D
Follow up:
Apparently the patent workes with mutants of kod and pfu that already have higher fidelity and kod in their system is less fidelous than pfu
In other publications lod is shown as more fidelous
useful information
I am yet to get my hands on the home-brewed pfu. However the commercially available pfu is giving amplification in NTC. Any clue?
I would start systematically start checking your reagents for contamination, happens in the blink of an eye, could be in your pfu buffer stock, could be in your primer stocks etc.
Anybody please post a 5X affordable GC enhancer recipe.
I tried your HF buffer recipe 1 and its working very well. Now I have the newly purified pfu and buffer to perform tons of pcrs haha. I hope the enzyme dont loose the activity for at least 6 months.
Ty for the very useful blog.
Sweet bro, glad it worked for you! Enjoy 🙂
My last batch lasted two years, the activity fell off sharply after that though. I’m playing with 10% trehalose in the storage buffer used to purify the enzyme, it can allegedly improve stability in the cold.
Oh, I didnt receive any notification of your answer. Sorry for replying 1 month later. Thank you for the sugestion. Im gonna try that. just one question: the buffer with 10% trehalose just the storage buffer, right? the last buffer in which we dialysis the sample from the IMAC?
We tried to freeze dry pfu-sso7d in 50 microliter 1x HF buffer with 5% trehalose. It works well after 1 month.
Anyone tried buffer #3? If yes please share the experience here.
Cool stuff here man im just confused which buffer ended up being the best? Also is there a THE pfusion construct circulating? Plus whats the deal with np40 and tween do we add from start of purification or only in final storage buffer?
Sorry for the confusion, I know it’s kind of a flow of conciousness vs a protocol. I’ve got another polymerase coming shortly so I’ll probably revamp the pfu protocol at the same time.
So best buffer for pfusso7d? Buffer #1 works well, however I’ve had users try it and show that commercial buffer outperforms it. So, use either commercial or Buffer #1. It’s a project I really want to tackle at some point, careful buffer optimization of this enzyme.
So Barrick’s pfusso7d plasmid is the most common circulating, however if you wanted an upgrade you could get a mutated clone synthesized for 150$ USD, theres probably a dozen mutations you could make. Although I’m told they have different preferences for buffers themselves.
Cheers mate keep up the great work
Also was looking into your other constructs. Does your RNAse inhibitor express Soluble in bl21 and hows the yield ? Does it have an mbp tag?
I haven’t announced it yet, but my pML-MRI is being replaced by MBP-Sumo-MRI and MBP-TEV-MRI, which was created by a friend of the blog, Ye Yang. The original plasmid produces a decent yield but you have to grow it at 16c…the MBP version is far superior in terms of soluble yield at more reasonable temperatures. I’ve been sending out the new clones, just haven’t done up a new post yet, feel free to shoot me an email.
Thats so cool i have a sumo protease construct i can send you some if you need. Also does the protease recognize the sumo in the middle?
My best guess for the GC enhancer from NEB is 25% DMSO.
According to it MSDS, the only components is “Trade Secret”. (10-30%) if we check the Europe version , it tell us the EC No. 200-664-3, which is DMSO.
Also the
ecotoxicity to plant shows like below
Algae/aquatic plants: EC50: 12350 – 25500mg/L (96h, Skeletonema costatum)
if we google this, we can find this in another NEB MSDS for competent cell.
Dimethly Sulfoxide 67-68-5
Algae/aquatic plants: EC50: 12350 – 25500mg/L (96h, Skeletonema costatum)
All other ecotoxicity to Fish or Crustacea are consistent between DMSO and the “Secret”.
Also, if we check the MSDS for Japan and Korean. the percentage shows 20-30%
It is 5x buffer. i would say the final concentration is 5%
Also i am trying to figure out the components in Q5 buffer. According to the MSDS of Q5 buffer for Korean
pH8.5
60-100% water
3-7% secret 1
1-5% KCl
1-5% NH4SO4
1-5% Tris
1-5% Secret 2
0.1-1% MgCl2
For the Secret 2, same to the Q5 GC enhancer, I would guess it is Tetramethylammonium chloride. According to paper (Nucleic Acids Res. 1995 Aug 25; 23(16): 3343–3344.), TMAC does enchance PCR reactions, and they suggest 60mM. According to Q5 master mix MSDS for Korean, the TMAC is 0.1-1 %, that is 2x? So i believe the the working concentration in Q5 buffer of TMAC is 60 mM
For the Secret 1. There is no ecotoxicity information, so it exclude most of the chemicals. Even water has this info. BSA doesn’t have the info. But 3-7% BSA, it is too much. It
I was stopped here…. any other clue?
have you ever tried the Taq expression construct from addgene(https://www.addgene.org/25712/), we have failed several times to induce its expression in BL21, no detectable expression was observed, and we totally have no idea where is the trick.
Hi, Bo!
Try DH5a strain.
Induce expression by adding IPTG to 0.5mM at A600 = 0.3-0.6 and incubate overnight at 37C.
Hope this would help.
Or you may try new Taq pol expression strain:
https://rokebio.com/products/instant-taq-stab-culture
I didn’t tried it by myself.
I found a very simple protocol without using column. just lyse the cells, centrifuge, heat, centrifuge and dialysis.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6635313/
gonna try it out.
if you have any chelating column or resin, I would suggest not to waste time on it. I don’t see a step to remove E.coli DNA. Our you could add 1.5% streptomycin sulfate to the lysate, that could precipitate out nucleic acid.
They use BugBuster for lysis, as far as I remember after lysing cells with BugBuster, the sample does not get viscous due to DNA (probably due to some propriety non-DNAse way?) so it is not simply boil’n’go.
My simplest protocol to this day is add lysozyme, boil, add PEI w/ 1M NaCl (precipitate DNA), dialyse. I do not have an exact protocol, did a trial run once to see if it works, and it worked. But still, when I am doing an actual prep I do the works, that is 70 C incubation followed by Ni-NTA followed by size exclusion.
Never waste pure thoughts on an impure protein 😀
ML
Thanks for pointing it out. I haven’t use the BugBuster before. I found this in their flyer ” Benzonase® Nuclease and rLysozyme™ Solution” .
Returning back to this after a year…
There are some guys who created a cell line containing Taq + LyseR from lambda + S. marcercens nucA (a.k.a. Benzonase), where they incorporated a operon structure involving LyseR / nucA / Apra resistance to yipD (?) phosphate-regulated operator. You can find the link here – https://www.biorxiv.org/content/10.1101/2021.09.25.461774v1.full.pdf
For generating lysates for cell-free expression, I got a LyseR plasmid from Addgene, where LyseR expression is driven through Amp promoter. Cells are viable and grow nicely, and you get nearly complete lysis when you use something like chloroform (or the paper above uses 0.1% Triton X-100 with freeze thaw, which also works nicely). I tried a similar system with nucA directed to periplasm with pelB secretion signal however the cell growth was a bit stunted so maybe I should try a weaker promoter? Who knows… But at the end you get a lysate that is completely cleared of nucleic acid contamination and lyses nicely.
Also some guys incorporated some salt bridge mutants to make nucA more heat labile, since the enzyme itself is quite heat stable. I did express other proteins using this system but did not dare to utilize this (the nucA portion) with polymerases and such because I don’t know I can completely clear nucA short of size exclusion.
For the other guys, I actually tried the protocol outlined here https://www.ncbi.nlm.nih.gov/pmc/articles/PMC307412/ with Taq, Phusion and KOD and produced working enzymes, albeit with significant nucleic acid contamination that you can see in the gel clearly (seems mostly like RNA though…) so you can opt out of ammonium sulfate, PEI and Ni-NTA and produce enzymes for daily use.
My 0.02$ 🙂
Take care,
ML
p.s. – Anyone who want to contact me can mail me at cihan.aydin at gmail
Hi MadLab
Could you please send me your pfu purification protocol. I am still getting DNA contamination in my purification. I have used Streptomycin Sulphate, DEAE, PEI and 1M NaCl wash but all of these failed to remove the DNA.
I don’t know how trace amount DNA you want. Typically the methods you mentioned should work okay. One thing i could point is using ammonium sulfate/sodium sulfate rather than NaCl in your buffer and washing is better to remove DNA. Because SO4 is more like PO4 than Cl.
If you need further remove DNA, i would suggest DNAase before heat shock. or apply pfu to Heparin column after Ni-NTA. Heparin is a nuclease acid analog. Wash with buffer contain 100 mM KCl or higher. pfu should be elute by buffer with 550 mM KCl maybe higher. you might need to optimize the conditions, like linear gradient to find the best wash and elute conditions. Or other cation column should works.
Good luck
Do you have a Q column lying around? Maybe try that?
Thanks to both of you. I have tried the q-sepharose column. I thought the protein would be in flow through or wash fraction but I didn’t elute with KCl. I will be try that.
One trick I picked up from a friend was to wash the polymerase with lysis buffer + 1M NaCl while the protein is still stuck to the Ni-NTA beads, washes out bound nucleic acids.
For taq/pfu, bind/wash/elute with nickle beads always seems to work well, however I’ve moved away from the phosphate buffer in the lysis buffer and changed to Tris. I think the phosphate can chelate ions that are important for PCR if you don’t get rid of it completely. I’ve had a bad run of purifications to what I think is phosphate…
Dear All,
did someone find a final solution to prevent rapid activity loss?
For our last purification we got a ton of working Phusion, however after roughly 2 weeks it’s not working anymore.
We are using dialysis after elution for the storage buffer. Additionally, I solved DTT freshly from powder before adding it before storing at -20 °C. Other chemicals, as NP-40 are of high quality (Sigma).
Until now I couldn’t figure out a way to recover the activity.
Hoping for helpful comments 🙂
Kind regards
Markus
I figured it out by now.
It seems like the Tween was badly distributed before storage at -80°C.
So now after taking out a Phusion aliquot, I add new Tween, Triton and DTT as if it was never added before. Now the Phusion stays active until the aliquot is empty.
Kind regards
Markus
hi mad lab and pipettejockey
i want to know what should i do to remove any DNase and RNAse afer Ni-colomn
and what is your protocol and buffers for it
thanks
Hi dear mad lab
thanks for your contents
do you have the specific activity of phusion for further calculation in concentration?
I recently determined that NEB LongAmp Buffer works better than any of the ‘home made’ Phusion buffer I’ve tried for the PfuI-Ssod7 I purified in 2017. (It is a clone without a his6 tag and the enzyme was purified by affinity chromatography on Sepharose SP). And fortunately, NEB actually provides the composition of that buffer
1X LongAmp® Taq Reaction Buffer Pack
60 mM Tris-SO4
20 mM (NH4)2SO4
2 mM MgSO4
3% Glycerol
0.06% IGEPAL® CA-630
0.05% Tween® 20
(pH 9.1 @ 25°C)
Hi. Thank you for such an informative and ever attractive community. Can i get an aliquot of the home brew phusion polymerase plasmid. I am from India. please let me know the possibility.